It seems we can’t find what you’re looking for. Perhaps searching can help.
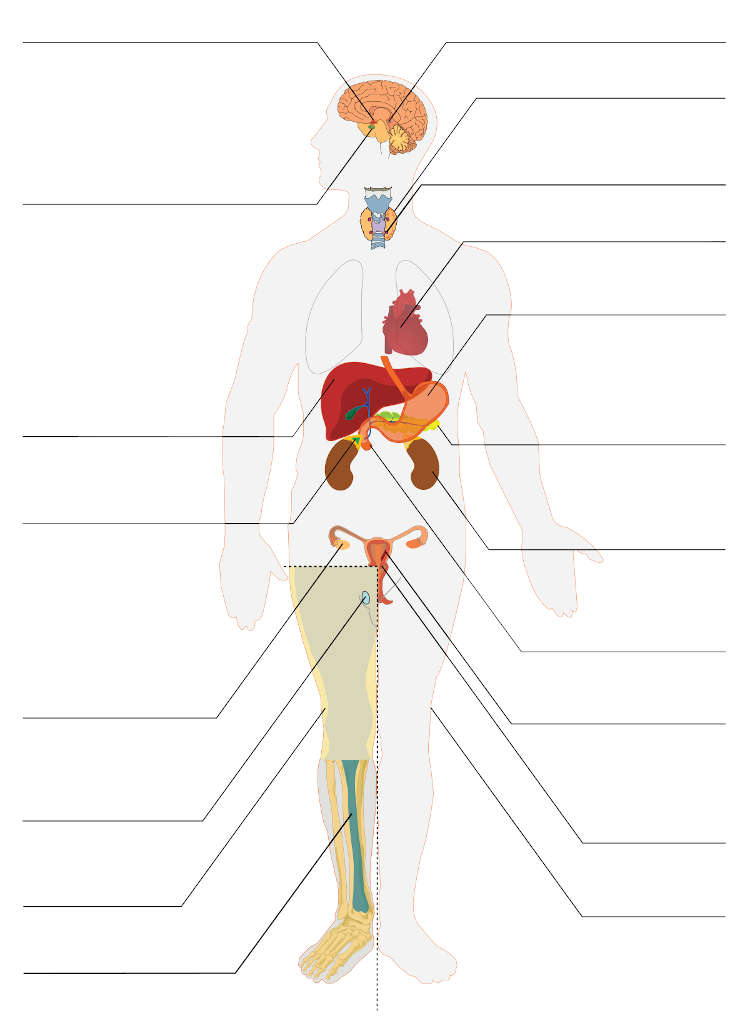
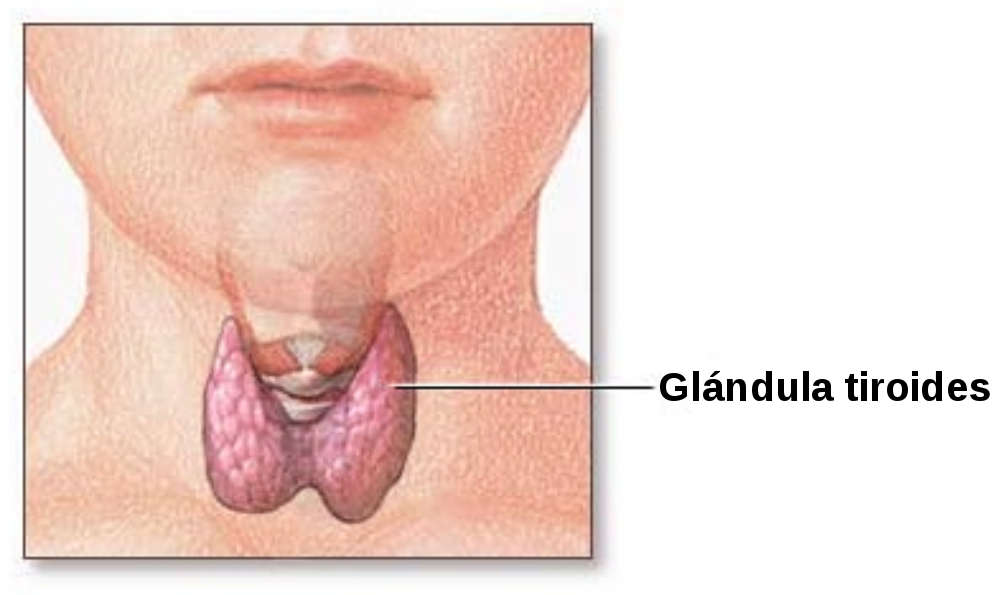
CBD Oil and Thyroid Medication
CBD Oil for Adrenal Fatigue
The Effects of Cannabinoids on the Endocrine System
Does CBD Increase Appetite?
CBD Oil for the Thyroid Gland
Copyright © 2024 | WordPress Theme by MH Themes